Rohan Shah
M.D./Ph.D. Student
The University of Chicago
Biography
I am an M.D./Ph.D. student at the University of Chicago studying chromatin epigenetics in the Ruthenburg Laboratory. My research interests include developing quantitative applications of common techniques in modern molecular biology. This includes designing methods for quantitative studies of chromatin immunoprecipitation and creating novel tools for analysis of next-generation sequencing data.
Download my curriculum vitae.
Interests
- Epigenetics
- Antibodies
- Chromatin immunoprecipitation
- Next-generation sequencing
Education
-
M.D. in Medicine (ongoing)
The University of Chicago Pritzker School of Medicine
-
Ph.D. in Genetics, Genomics, and Systems Biology, 2022
The University of Chicago
-
B.S. in Biological Chemistry with Honors, 2018
The University of Chicago
Skills
Windows
Ubuntu
Statistics
R
Microsoft Excel
Adobe Illustrator
Research Experience
Responsibilities include:
- Developing a novel form of chromatin immunoprecipitation to quantify internal histone modifications
- Developing methods for Bayesian reallocation of ambiguously mapped next-generation sequencing reads
- Serving as systems administrator to manage and maintain lab servers and promote integration of computational resources with ongoing research projects throughout the lab
- Working in multiple ongoing collaborations on the roles of histone modifications as epigenetic regulators
- Worked as part of multiple ongoing research projects with Dr. Renslow Sherer at the University of Chicago.
- Performed chart abstractions of first 414 COVID-19 admissions to the University of Chicago Medical Center
- Worked on subgroup study examining risk factors and outcomes of COVID-19 patients with coinfections
Responsibilities included:
- Training researchers on-site in methods and principles of calibrated chromatin immunoprecipitation
- Consulting on feasibility of simplification and consolidation of materials and proecdures for commercialization
- Assisting with preparing documentation and troubleshooting guides for calibrated chromatin immunoprecipitation kit
Teaching Experience
Honors and Awards
Best Talk, UChicago Molecular Biosciences Retreat
Ting-Wa Wong MD, PhD Scholarship
Growth, Development and Disabilities Training Program Appointee
Summer Research Program Overall Excellence in Basic Sciences
Keystone Symposia Scholarship Awardee
Frances E. Knock Prize for Outstanding Academic Achievement in Chemistry
Phi Beta Kappa Honor Society, Chapter Beta of Illinois
University of Chicago Student Marshal
Katen Scholar
Beatrice G. and Nate H. Sherman Fellowship
Featured Publications
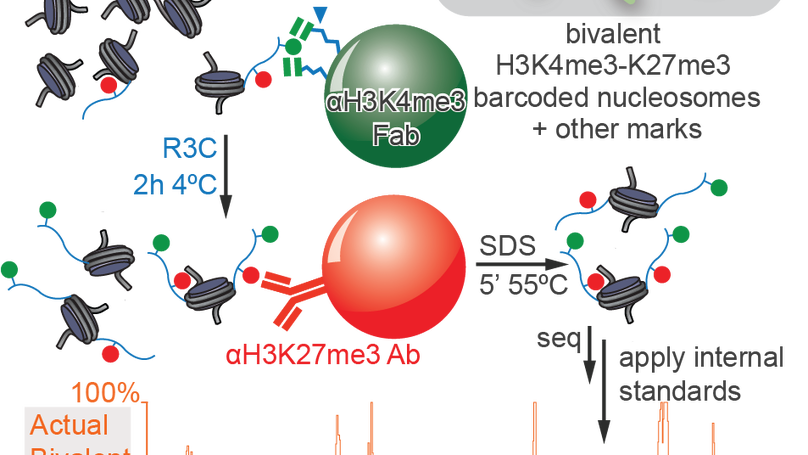
Re-evaluating the role of nucleosomal bivalency in early development
Nucleosomes, composed of DNA and histone proteins, represent the fundamental repeating unit of the eukaryotic genome; posttranslational modifications of these histone proteins influence the activity of the associated genomic regions to regulate cell identity. Traditionally, trimethylation of histone 3-lysine 4 (H3K4me3) is associated with transcriptional initiation, whereas trimethylation of H3K27 (H3K27me3) is considered transcriptionally repressive. The apparent juxtaposition of these opposing marks, termed “bivalent domains”, was proposed to specifically demarcate a small set of transcriptionally-poised lineage-commitment genes that resolve to one constituent modification through differentiation, thereby determining transcriptional status. Since then, many thousands of studies have canonized the bivalency model as a chromatin hallmark of development in many cell types. However, these conclusions are largely based on chromatin immunoprecipitations (ChIP) with significant methodological problems hampering their interpretation. Absent direct quantitative measurements, it has been difficult to evaluate the strength of the bivalency model. Here, we present reICeChIP, a calibrated sequential ChIP method to quantitatively measure H3K4me3/H3K27me3 bivalency genome-wide, addressing the limitations of prior measurements. With reICeChIP, we profile bivalency through the differentiation paradigm that first established this model: from naïve mouse embryonic stem cells (mESCs) into neuronal progenitor cells (NPCs). Our results cast doubt on every aspect of the bivalency model; in this context, we find that bivalency is widespread, does not resolve with differentiation, and is neither sensitive nor specific for identifying poised developmental genes or gene expression status more broadly. Our findings caution against interpreting bivalent domains as specific markers of developmentally poised genes.
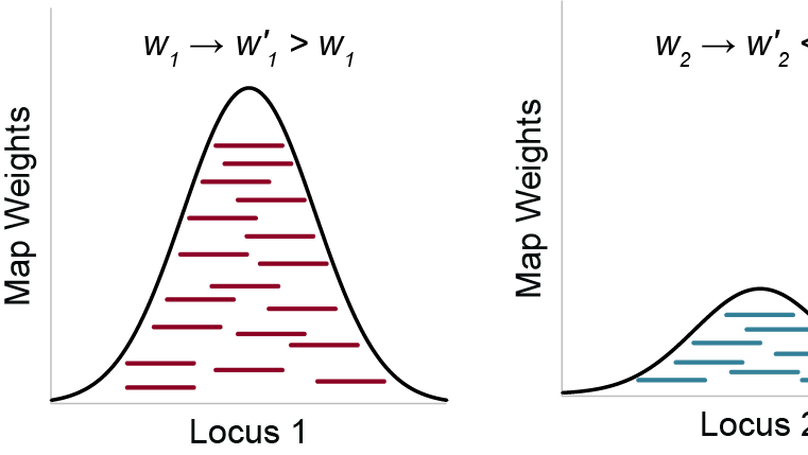
Sequence deeper without sequencing more: Bayesian resolution of ambiguously mapped reads
Next-generation sequencing allows researchers to efficiently determine the sequences of hundreds of millions of short DNA fragments from an experiment. To find the origins of those fragments, the corresponding sequences are aligned to the genome; these alignments can then be used in downstream analyses. However, this alignment process is complicated by the fact that the genome has many highly similar and repetitive sequences, making it difficult or impossible to unambiguously assign some sequences to a single genomic location. To address this problem, we have developed SmartMap, which serves to process and appropriately weight the alignments of reads that map to more than one genomic location. This enables us to examine many genomic regions that were previously “invisible” to analysis and helps us draw new insights into the regulation and function of repetitive elements of the genome.
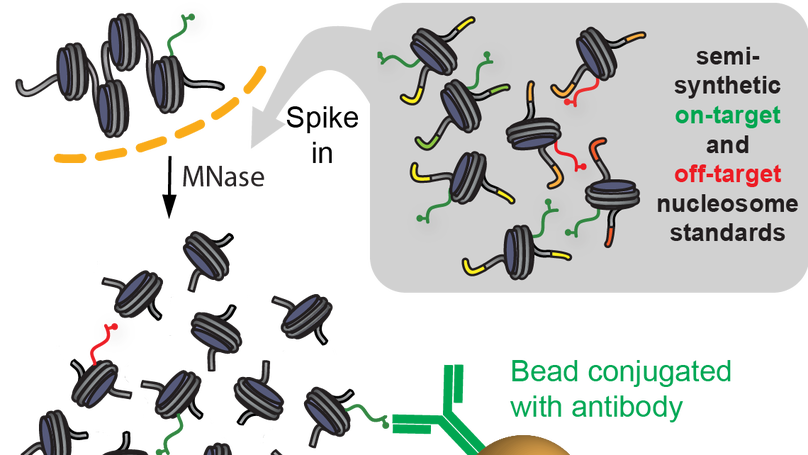
Native internally calibrated chromatin immunoprecipitation for quantitative studies of histone post-translational modifications
Chromatin immunoprecipitation coupled to next-generation sequencing (ChIP-seq) has served as the central method for the study of histone modifications for the past decade. Here we provide a detailed protocol for internally calibrated ChIP (ICeChIP), a method we recently developed to resolve these problems by spike-in of defined nucleosomal standards within a ChIP procedure.
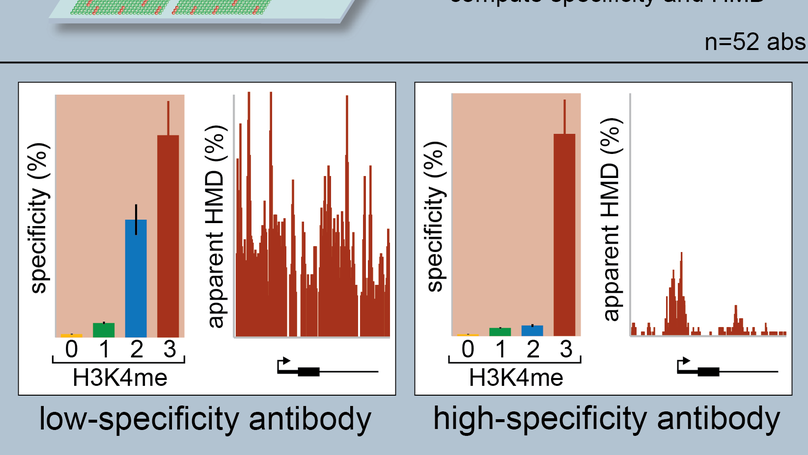
Examining the Roles of H3K4 Methylation States with Systematically Characterized Antibodies
Histone post-translational modifications (PTMs) are important genomic regulators often studied by chromatin immunoprecipitation (ChIP), whereby their locations and relative abundance are inferred by antibody capture of nucleosomes and associated DNA. Here, we use histone peptide arrays and internally calibrated ChIP (ICeChIP) to characterize 52 commercial antibodies purported to distinguish the H3K4 methylforms (me1, me2, and me3, with each ascribed distinct biological functions).
All Publications
Contact
- rohan.n.shah@outlook.com
- 773 702 2556
- 920 E. 58th Street, Ruthenburg Lab, Chicago, IL 60637